
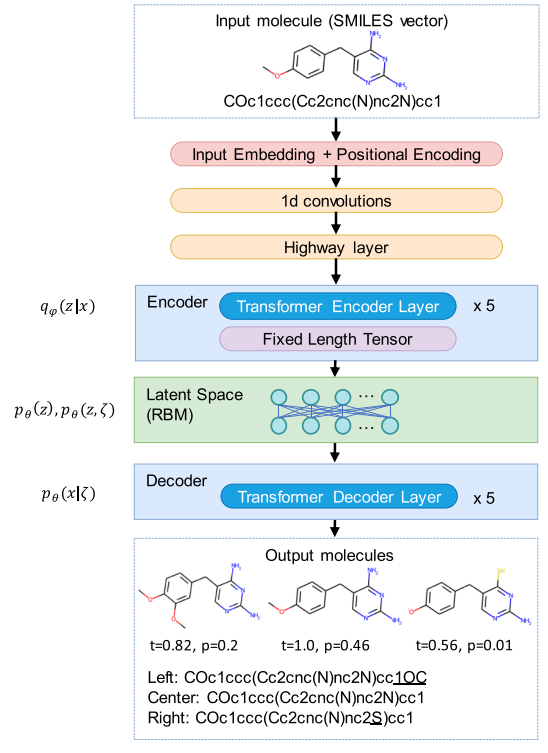
شرکت Gero، فعال در بیوتکنولوژی مبتنی بر هوش مصنوعی با تمرکز بر پیری و طول عمر، امکان استفاده از محاسبات کوانتومی برای طراحی دارو و شیمی مولد را نشان داده است، که اکنون نوید قابل توجهی را برای آینده مراقبت های بهداشتی ارائه می دهد. این تحقیق که در Scientific Reports منتشر شده است، چگونگی استفاده از یک مدل یادگیری ماشینی ترکیبی کوانتومی-کلاسیک را برای ارتباط بین دستگاههای محاسباتی کلاسیک و کوانتومی با هدف تولید ساختارهای شیمیایی جدید برای داروهای بالقوه - ابتدا یک صنعت - مورد استفاده قرار میدهد.
این مقاله تحقیقاتی پس از پیشرفتهای اخیر Gero دنبال میشود، که بحثهای شدیدی را در میان متخصصان طول عمر در جامعه علمی به راه انداخت، مقاله ای که ادعا میکرد انسانها میتوانند پیری را متوقف کنند - اما نه به طور کامل. در اوایل سال جاری، Gero یک قرارداد کشف هدف با Pfizer اعلام کرد که به موجب آن پلتفرم فناوری یادگیری ماشینی Gero برای کشف اهداف درمانی بالقوه برای بیماریهای فیبروتیک با استفاده از دادههای انسانی در مقیاس بزرگ استفاده میشود. در این خط تحقیقاتی جدید، این تیم بررسی کرد که آیا یک سیستم هوش مصنوعی مولد ترکیبی - یک شبکه عصبی عمیق که در ارتباط با سختافزار کوانتومی موجود تجاری کار میکند - میتواند ساختارهای شیمیایی منحصربهفردی را پیشنهاد کند که از نظر مصنوعی امکانپذیر بوده و دارای خواص مشابه دارو هستند.
این چشمانداز مولکولی میتواند کلید درمانهای اساسی در آینده برای بیماریهای صعبالعلاج مرتبط با افزایش سن و خود پیری را در خود جای دهد. با این حال، اندازه و پیچیدگی این فضای تنوع شیمیایی ناشناخته نیازمند ابزارهای نوآورانه برای انتخاب مولکولهای جدید، فعال بیولوژیکی و در عین حال قابل دسترس مصنوعی است که منتظر تبدیل شدن به داروهای آینده هستند. طراحی دارو در تقاطع قلمرو پدیده های کلاسیک و کوانتومی عمل می کند و نیاز به تعیین همزمان خواص کوانتومی مولکول های دارو مانند و اثرات آنها بر سیستم های زنده توصیف شده توسط فیزیک کلاسیک دارد. به همین دلیل است که محاسبات کوانتومی به طور قابل توجهی ظرفیت ما را افزایش می دهد. برای چالشبرانگیزترین بیماریها و شرایط، از جمله خود پیری، درمانهای متحول کننده ایجاد کنید.
محققان یک مدل ترکیبی ایجاد کردند که یک رمزگذار خودکار تغییرات گسسته فشرده (DVAE، یک الگوریتم شیمی مولد) را به شکلی ترکیب میکند که میتواند در دستگاه کوانتومی پیشرفته ای به نام آنیل کوانتومی D-Wave پیاده سازی شود. سیستم پیشنهادی یک حالت مولد کوانتومی/کلاسیک ترکیبی است که برای نمونهبرداری از توزیع مولکولهای دارو مانند و مصنوعی در دسترس آموزش دیده است. هنگامی که آموزش کامل شد، سیستم می تواند در حالت مولد اجرا شود و 2331 ساختار شیمیایی جدید با خواص معمول برای ترکیبات فعال بیولوژیکی را پیشنهاد کرد. به طور دلگرم کننده، کمتر از 1٪ از مولکول های تولید شده شباهت زیادی به هر مولکولی در مجموعه آموزشی داشتند که نشان دهنده سطح بالایی از تازگی در ترکیبات تولید شده است.
توسعه الگوریتم های کوانتومی و مدل های ترکیبی کوانتومی-کلاسیک یادگیری ماشینی برای کشف دارو می تواند به طور قابل توجهی زمینه شیمی دارویی را پیشرفت دهد. از آنجایی که وسعت فضای ساختاری مولکولهای احتمالی شبه دارو، چالش مهمی برای محاسبات کلاسیک ایجاد میکند، محاسبات کوانتومی ممکن است رویکرد بسیار کارآمدتری ارائه دهد.
همانطور که سخت افزار کوانتومی بالغ می شود، اجزای خاص شبکه می توانند به همتای کاملا کوانتومی خود تبدیل شوند و به طور بالقوه سیستم را به همتای کوانتومی اش تبدیل می کنند که می تواند از توزیع های غنی تر و غیر کلاسیک نمونه برداری کند. این در نهایت می تواند آموزش سیستم را تسریع کند و به طور بالقوه مدل های مولد تقویت شده کوانتومی را برای کاربردهای طراحی دارو کارآمدتر کند.
با قویتر شدن رایانههای کوانتومی، ما انتظار داریم که در مطالعات مختلف، بهویژه در حوزه یادگیری ماشینی که برای مشکلات مکانیکی کوانتومی بهطور طبیعی اعمال میشود، بیشتر و بیشتر مفید واقع شوند. در پنج تا ده سال آینده، ما شاهد یک روش جدید در تولید داروها و مواد ایجاد شده با کمک کامپیوترهای کوانتومی خواهیم بود.
لینک مقاله:
https://www.nature.com/articles/s41598-023-32703-4










